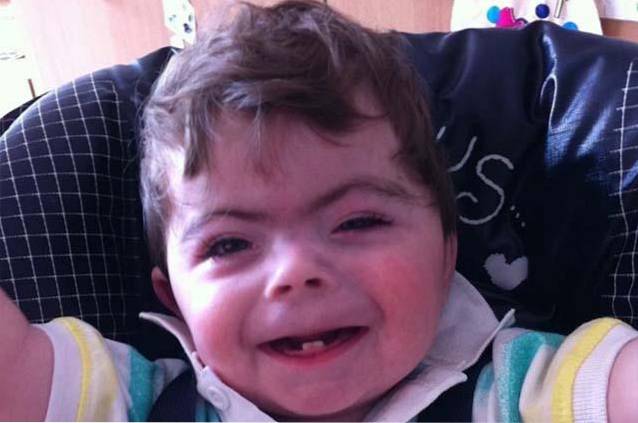
Sindromul Cornelia de Lange este o patologie de origine genetică care se caracterizează prin prezența unei întârzieri cognitive semnificative însoțite de diverse trăsături fizice malformative.
La nivel clinic, se observă trei cursuri clinice diferențiale: severă, moderată și ușoară. Semnele și simptomele sunt de obicei constituite de o configurație facială atipică, de malformații musculo-scheletale și de o dezvoltare cognitivă și psihomotorie întârziată. În plus, se pot distinge alte tipuri de anomalii legate de malformații cardiace, pulmonare și / sau digestive..
În ceea ce privește originea sindromului Cornelia de Lange, etiologia sa a fost legată de prezența mutațiilor specifice în genele SMC3, SMC1A, NIPBL, printre altele. Diagnosticul este fundamental clinic, realizat pe baza caracteristicilor fizice și cognitive. Cu toate acestea, este însoțit de obicei de un test genetic de confirmare.
Tratamentul este orientat spre detectarea și tratamentul complicațiilor medicale. Medicale, logopedie, intervenția neuropsihologică și educația specială sunt esențiale.
Indice articol
Acest sindrom a fost descris inițial de Dr. Cornelia de Lange în 1933. Cercetările sale s-au bazat pe studiul a doi pacienți cu vârste cuprinse între 6 și 17 luni. Tabloul său clinic a fost caracterizat de o întârziere severă a creșterii fizice și a dezvoltării intelectuale asociate cu diferite trăsături malformative..
Având în vedere similaritatea ambelor cazuri, primul raport clinic despre această patologie a presupus existența unei cauze etiologice comune și publice.
Anterior, Brachmann (1916) reușise să publice câteva date de autopsie ale unui pacient în vârstă de copil cu unele caracteristici compatibile cu sindromul Cornelia de Lange..
În prezent, tabloul clinic al acestui sindrom a fost clasificat în trei fenotipuri diferențiale: severă, moderată și ușoară..
Sindromul Cornelia de Lange este o afecțiune genetică rară de natură congenitală, adică caracteristicile sale clinice sunt evidente încă de la naștere. Este definită ca fiind o boală multisistemică cu simptome asociate cu dezvoltarea fizică și cognitivă întârziată, malformații cranio-faciale sau malformații musculo-scheletice..
Deși evoluția clinică și severitatea acestui sindrom pot varia considerabil în rândul celor afectați, este o boală cu o rată ridicată a mortalității.
Persoanele cu sindrom Cornelia de Lange se caracterizează prin prezența unei configurații faciale atipice sau caracteristice și o întârziere în creșterea / dezvoltarea pre și postnatală..
Cel mai frecvent este că apar probleme de învățare, întârzierea dobândirii limbajului sau mersului și anomalii comportamentale.
Sindromul Cornelia de Lange este o boală rară în populația generală, de obicei este clasificată în cadrul bolilor rare. Datele epidemiologice nu sunt cunoscute exact. Incidența sa a fost estimată la un caz la 10.000-30.000 de nașteri..
Până în prezent, putem găsi peste 400 de cazuri diferite de sindrom Cornelia de Lange descrise în literatura medicală și experimentală..
Este o patologie care poate afecta ambele sexe în număr egal. Unii autori precum Gutiérrez Fernández și Pacheco Cumani (2016) sugerează o ușoară predominanță față de femei, cu un raport de 1,3 / 1.
În raport cu restul factorilor sociodemografici, cercetările actuale nu au identificat o prevalență diferențială asociată cu anumite țări sau grupuri etnice și / sau rasiale..
O bună parte a cazurilor diagnosticate sunt sporadice, deși mai multe familii afectate au fost identificate cu un model clar de moștenire dominantă..
Semnele și simptomele sindromului Cornelia de Lange se caracterizează prin tiparul lor larg de implicare.
Această boală este definită de prezența trăsăturilor faciale caracteristice, malformații musculo-scheletice la nivelul membrelor superioare și inferioare, întârzierea generalizată a creșterii pre și postnatale, împreună cu dezvoltarea altor anomalii fizice..
În continuare, vom descrie unele dintre cele mai frecvente caracteristici clinice în sindromul Cornelia de Lange:
La mai mult de 90% dintre cei afectați de sindromul Cornelia Lange, este posibil să se identifice o întârziere a dezvoltării fizice sau a hipogreșterii globale. Creșterea este de obicei afectată atât prenatal, cât și postnatal.
Cele mai frecvente caracteristici la nou-născuți sunt:
Aceste condiții durează de obicei până la maturitate. În acesta, se poate distinge o creștere situată sub cea așteptată pentru sexul și vârsta biologică a persoanei afectate.
Odată cu acest tip de modificări, pot fi identificate unele anomalii legate de hrănire. Dificultățile de a înghiți sau de a mesteca alimente sunt frecvente în primele etape ale vieții.
Combinarea alterărilor craniene și faciale are ca rezultat dezvoltarea unui fenotip facial caracteristic la persoanele care suferă de sindromul Cornelia de Lange..
Unele dintre cele mai frecvente anomalii includ:
Întârzierea dezvoltării cognitive și psihomotorii constituie una dintre constatările clinice centrale în sindromul Cornelia Lange. De obicei se identifică o dobândire lentă a abilităților legate de activitatea motorie sau mentală.
Cele mai afectate repere sunt achiziționarea șezutului, zâmbetul afectiv, gâlgâitul, mișcarea independentă, emisia primelor cuvinte, înțelegerea și ordinele, hrănirea, ambulanța sau toaleta independentă.
La mulți dintre cei afectați, poate fi identificat un IQ mediu asociat cu dizabilități intelectuale ușoare sau moderate.
Comportamentul celor afectați de sindromul Cornelia de Lange prezintă de obicei câteva trăsături distinctive:
Sindromul Cornelia de Lange este, de asemenea, asociat cu dezvoltarea diverselor complicații medicale.
Cele mai frecvente cauze de deces sau agravare a stării medicale a celor afectați sunt legate de:
Variabilitatea semnelor și simptomelor sindromului Cornelia de Lange a permis o clasificare a evoluției sale clinice:
De obicei este cel mai grav. Modificările și anomaliile se caracterizează prin prezența subprodusului fizic, malformații musculo-scheletice, trăsături faciale anormale, limitarea mobilității articulare, întârziere cognitivă și alte complicații medicale (auditive, oculare, digestive, reno-urologice, cardiace și genitale)..
În acest subtip, modificările fizice nu sunt de obicei foarte evidente, în special la nivelul extremităților. Cei afectați nu au de obicei deficite intelectuale grave. Cel mai frecvent este că diagnosticul se face dincolo de stadiul neonatal.
Cursul său clinic este caracterizat fundamental de variabilitatea clinică. Caracteristicile feței sunt prezente în majoritatea cazurilor, dar expresia restului anomaliilor este variabilă..
Originea sindromului Cornelia Lange este asociată cu prezența anomaliilor genetice. În cazurile examinate, a fost posibilă identificarea mutațiilor specifice în 5 gene diferite: NIPBL, SMC1A, HDAC8, RAD21 și SMC3.
Cea mai frecventă modificare este legată de gena NIPBL, identificată la mai mult de jumătate dintre cei afectați. Restul anomaliilor genetice sunt mai puțin frecvente.
Toate aceste gene joacă un rol proeminent în producerea proteinelor legate de complexul de coezină, responsabile de reglarea structurii și organizării cromozomiale, stabilizarea informațiilor genetice în celule și repararea ADN-ului..
În plus, ele îndeplinesc, de asemenea, mai multe funcții fundamentale în dezvoltarea prenatală a extremităților, feței și a altor regiuni și a sistemelor corpului.
Diagnosticul sindromului Cornelia de Lange este clinic. În prezent nu există niciun test de laborator care să indice definitiv prezența acestuia. În zona medicală, cel mai frecvent este utilizarea criteriilor de diagnostic propuse de Kline și colab..
Acestea se referă la identificarea anomaliilor cranio-faciale, în creștere și dezvoltare, în extremități, alterări neurosenzoriale și cutanate, tulburări de comportament etc..
În plus, este important să se efectueze o analiză genetică moleculară pentru a identifica prezența mutațiilor asociate cu sindromul Cornelia de Lange..
Deși nu există nici un remediu pentru sindromul Cornelia de Lange, abordarea sa terapeutică implică proiectarea unei urmăriri medicale continue împreună cu tratamentul complicațiilor..
Autorii Gil, Ribate și Ramos (2010) subliniază unele dintre cele mai utilizate abordări.


Nimeni nu a comentat acest articol încă.