
Sindromul Prader-Willi (SPW) este o patologie multisistemică care are o origine genetică congenitală. Este o boală complexă care afectează apetitul, creșterea, metabolismul, comportamentul și / sau funcția cognitivă.
La nivel clinic, în etapa copilăriei, această boală se caracterizează prin prezența diferitelor descoperiri medicale, cum ar fi slăbiciune musculară, tulburări de alimentație sau întârziere generalizată a dezvoltării..
În plus, la nivel cognitiv și comportamental, o bună parte a persoanelor afectate de sindromul Prader-Willi prezintă o afectare intelectuală moderată sau o întârziere, care este însoțită de diverse probleme de învățare și comportament..
Deși sindromul Prader-Willi este considerat o boală rară sau neobișnuită, numeroase studii indică faptul că este una dintre cele mai frecvente patologii din zona genetică. Diagnosticul acestei boli se face în principal pe baza constatărilor clinice și a testelor genetice complementare..
În ceea ce privește tratamentul, un remediu pentru sindromul Prader-Willi nu a fost încă identificat, astfel încât abordarea terapeutică este orientată spre tratarea simptomelor și complicațiilor, obezitatea fiind constatarea medicală care reprezintă cea mai mare amenințare pentru cei afectați.
Astfel, în raport cu prognosticul și calitatea vieții, ambele vor depinde de gravitatea problemelor medicale asociate și de tulburările de comportament sau cognitive care se pot dezvolta..
Indice articol
Diferite rapoarte clinice indică faptul că sindromul Prader-Willi (PWS) a fost descris inițial de J. L. Down, în 1887, după diagnosticarea unuia dintre pacienții săi cu „polisarcia”..
Cu toate acestea, doctorii Prader, Labhart și Willi au fost cei care, în 1956, au descris alte 9 cazuri și au dat numele acestei patologii. În plus, caracteristicile și criteriile de diagnostic ale sindromului Prader-Willi au fost sistematizate de Holm și colab..
Sindromul Prader-Willi este o alterare genetică congenitală, adică este o patologie care este prezentă din momentul nașterii și va afecta individul de-a lungul vieții sale dacă nu există o intervenție terapeutică curativă..
Această patologie prezintă un curs clinic complex, caracterizat prin numeroase manifestări medicale.
Deși astăzi fenotipul sindromului Prader-Willi este mai precis cunoscut, a fost în ultimii 25 de ani, când s-au înregistrat progrese semnificative în analiza și înțelegerea acestei boli..
Expresia sindromului Prader-Willis este diversă, tinde să afecteze mai multe sisteme și structuri, majoritatea modificărilor fiind legate de disfuncția hipotalamică.
Hipotalamusul este o structură neurologică care are un rol esențial în controlul funcțiilor homeostatice: reglarea foamei, setei, ciclurile somn-veghe sau reglarea temperaturii corpului.
În plus, hipotalamusul eliberează diferiți hormoni în diferite glande: creștere, sexual, tiroidă etc..
În cele din urmă, trebuie să subliniem că sindromul Prader-Willis poate apărea, de asemenea, în referință în literatura medicală și experimentală cu alți termeni precum sindromul Prader-Labhart-Willi sau cu acronimul PWS..
De asemenea, alte sinonime sunt sindromul Labhart Willi, sindromul Praser Labhart Willi Fancone sau sindromul de distrofie hipogenitală..
Sindromul Prader-Willi (PWS) este o boală genetică rară. Termenul de boală rară (ER) este utilizat pentru a se referi la acele patologii rare sau puțini oameni care suferă de aceasta.
În prezent, se estimează că sindromul Prader-Willi este o patologie cu o frecvență aproximativă de 1 caz la 10.000-30.000 de oameni din întreaga lume..
Pe de altă parte, în ceea ce privește distribuția pe sexe, sa observat că această patologie afectează în mod egal bărbații și femeile și nu este asociată cu grupuri etnice sau regiuni geografice..
În plus, sindromul Prader-Willi este considerat principala cauză a obezității de origine genetică.
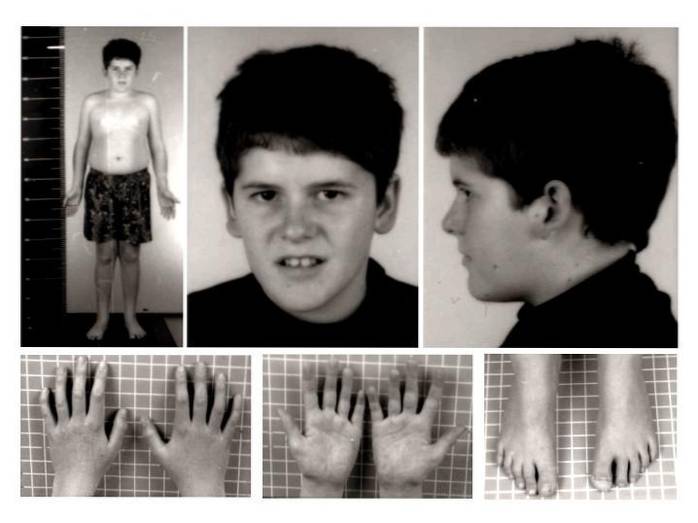
La nivel clinic, sindromul Prader-Willi a fost în mod tradițional asociat cu hipotonie neonatală, hipogonadism, hiperfagie, obezitate, statură scurtă, întârziere generalizată în dezvoltare, dizabilitate intelectuală moderată, aspect facial atipic și modificări comportamentale diferite..
În ciuda acestui fapt, expresia clinică a acestei patologii este foarte eterogenă și variază semnificativ în rândul persoanelor afectate..
În plus, semnele și simptomele caracteristice ale sindromului Prader-Willi tind să varieze în funcție de dezvoltarea biologică, deci putem observa diferite constatări clinice în perioada fetală și neonatală, perioada copilăriei sau a copilăriei timpurii, etapa școlară și, în final, etapa adolescentă.
Într-un mod sistematic, José A. del Barrio del Campo și colaboratorii descriu în detaliu cele mai caracteristice modificări din aria biomedicală, psihomotorie, cognitivă și comportamentală:
Cele mai caracteristice semne și simptome fizice includ tulburări precum; hipotonie, malformații sau deformări musculo-scheletice, greutate și înălțime reduse sau scăzute, pofta de mâncare în exces, obezitate, hipogonadism, tulburări de somn, tulburări respiratorii, trăsături ușoare atipice, alterarea reglării temperaturii corpului, printre altele.
Prezența sau dezvoltarea tonusului muscular redus. Flaciditatea musculară în această patologie se accentuează în special la nivelul gâtului și trunchiului, în special în stadiul neonatal și în primele luni de viață. Astfel, odată cu dezvoltarea biologică, tonusul muscular tinde să se îmbunătățească.
În acest caz, este obișnuit să se observe dezvoltarea scoliozei sau devierea coloanei vertebrale, alinierea slabă a membrelor inferioare (genu valgus) sau prezența picioarelor plate..
În plus, pot fi observate și alte tipuri de anomalii congenitale, cum ar fi reducerea dimensiunii picioarelor și a mâinilor, displazia șoldului, prezența a șase degete, printre altele..
Mai ales la momentul nașterii, atât înălțimea cât și greutatea copilului afectat sunt mai mici decât se aștepta pentru dezvoltarea și sexul lor. În ciuda faptului că valorile standard pot fi atinse la vârsta adultă, rata lentă de creștere tinde să modifice valorile adulte ale înălțimii și greutății.
Este obișnuit să observăm la persoanele cu sindrom Prader-Willi un pofta de mâncare nesățioasă, caracterizată printr-o obsesie sau fixare asupra alimentelor. Datorită aportului de cantități mari de alimente, cei afectați tind să dezvolte obezitate și alte complicații medicale asociate, cum ar fi diabetul zaharat de tip II.
Este frecventă și prezența alterărilor genitale. În mod specific, hipogonadismul sau dezvoltarea parțială a organelor genitale externe sunt foarte frecvente. În majoritatea cazurilor, dezvoltarea pubertală nu reușește să atingă stadiul final sau adult.
Sforaitul, frecvența crescută sau stopul respirator apar adesea recurent în timpul fazelor de somn. Astfel, cei afectați tind să prezinte diverse modificări legate de fragmentare, întârzierea somnului sau prezența trezirilor periodice.
Anomaliile și malformațiile musculo-scheletice pot afecta, de asemenea, caracteristicile cranio-faciale. Este posibil să se observe un craniu îngust, strabism ocular, piele și păr slab pigmentate, gură mică și buze subțiri, malformații dentare etc..
Persoanele afectate de sindromul Prader-Willi au de obicei probleme legate de reglarea temperaturii corpului, iar o altă constatare semnificativă este rezistența ridicată la durere.
Datorită prezenței malformațiilor musculo-scheletice și a tonusului muscular redus, dezvoltarea psihomotorie va fi mai lentă, afectând toate zonele.
Cei afectați prezintă de obicei dificultăți de serie pentru a desfășura orice tip de activitate care necesită una sau mai multe execuții motorii.
În ceea ce privește limitările cognitive, majoritatea celor afectați au o dizabilitate intelectuală ușoară sau moderată.
În plus, acestea prezintă de obicei unele zone specifice mai afectate, cum ar fi procesarea secvențială a informațiilor, memoria recentă sau pe termen scurt, rezolvarea problemelor aritmetice, procesarea auditivă a informațiilor verbale, modificarea atenției și concentrării și prezența rigidității cognitive..
Pe de altă parte, limbajul este un alt domeniu care este semnificativ afectat la persoanele cu sindrom Prader-Willi. De obicei se observă întârzieri în dobândirea abilităților fonologice, vocabular slab, modificarea construcției gramaticale, printre altele..
Problemele comportamentale și modificările sunt o altă descoperire tipică care poate fi observată în sindromul Prader-Willi, acestea variază în mod normal în funcție de vârstă sau stadiul de maturare în care se află persoana afectată, cu toate acestea, unele dintre cele mai frecvente trăsături comportamentale sunt:
Diverse investigații actuale au arătat că alterarea comportamentală tinde să crească odată cu vârsta și, prin urmare, tind să se înrăutățească, afectând zonele sociale, familiale și emoționale în mod generalizat.
După cum am subliniat în mai multe secțiuni de mai sus, sindromul Prader-Willi are o origine genetică.
Deși există în prezent o mare controversă cu privire la genele specifice responsabile de această patologie, toate datele arată că alterarea etiologică este localizată pe cromozomul 15.
De-a lungul studiului genetic al acestei patologii, au existat mai multe contribuții. Burtler și Palmer (1838) au detectat prezența anomaliilor în brațul lung al cromozomului 15 de la părintele patern, în timp ce Nicholls (1989) a observat că în alte cazuri tulburarea a fost legată de modificările cromozomiale ale mamei (Rosell-Raga, 2003).
În afară de aceasta, cea mai acceptată teorie despre originea acestei patologii este pierderea sau inactivarea diferitelor gene de expresie paternă care se află în regiunea 15q11-13 a cromozomului 15.
Diagnosticul sindromului Prader-Willi are două componente de bază, analiza constatărilor clinice și testarea genetică..
În ceea ce privește detectarea semnelor și simptomelor indicatoare, atât la copii, cât și la copiii mai mari, va fi esențial să se efectueze un istoric medical detaliat, individual și familial. De asemenea, este esențial să efectuați un examen fizic și neurologic..
Dacă pe baza acestor proceduri, există o suspiciune de diagnostic, va fi necesar să se prescrie diferite teste complementare pentru a determina prezența alterărilor și anomaliilor genetice..
Mai exact, aproximativ 90% din cazuri sunt diagnosticate definitiv prin teste de metilare a ADN-ului și alte teste suplimentare..
În plus, este, de asemenea, posibil să se pună un diagnostic prenatal al acestei afecțiuni, în principal la familiile cu antecedente de sindrom Prader-Willi..
Mai exact, testul de amniocenteză permite extragerea probelor de embrioni pentru efectuarea testelor genetice pertinente.
În prezent nu există nici un remediu pentru sindromul Prader-Willi. Ca și în alte boli rare, tratamentele se limitează la controlul simptomelor și îmbunătățirea calității vieții persoanelor afectate.
Cu toate acestea, unul dintre aspectele fundamentale va fi controlul nutrițional și al dietei, deoarece obezitatea este principala cauză a morbidității și mortalității în această patologie..
Pe de altă parte, prezența modificărilor cognitive și comportamentale va necesita intervenția profesioniștilor specializați atât în reabilitarea cognitivă, cât și în gestionarea tulburărilor de conduită..


Nimeni nu a comentat acest articol încă.