
Sindromul Waardenburg (SW) este o patologie de origine genetică clasificată ca tip de neuropatie. Caracteristicile sale clinice sunt definite de prezența surdității sau a pierderii auzului, a pigmentării anormale a ochilor, a părului sau a pielii și a diferitelor modificări ale feței..
Această patologie se caracterizează prin variabilitatea sa simptomatică largă, motiv pentru care se disting mai multe tipuri: tip I, tip II, tip III (sindrom Klein-Waardenburg sau psudo Waardenburg) și tip IV.

La nivel etiologic, sindromul Waardenburg are un tipar de moștenire autozomal dominant. Este de obicei asociat cu mutații specifice în genele EDN3, EDNRB, PAX3, SOX10, SNAI2 și MIT..
Diagnosticul se face pe baza diferitelor criterii clinice majore și minore. Cu toate acestea, este necesar să se efectueze diferite teste de laborator complementare. Nu există un tratament sau tratament specific pentru sindromul Waardenburg..
Intervenția cu această patologie se concentrează de obicei pe tratamentul tulburărilor de auz (proceduri chirurgicale, implanturi cohleare etc.), logopedie și reabilitare neuropsihologică, precum și psihologice.
Indice articol
Acest sindrom a fost descris inițial de geneticianul și oftalmologul olandez Petrus Johannes Waardenburg în 1848. În raportul său clinic a făcut referire la principalele caracteristici clinice:
Analizele ulterioare au identificat o variabilitate clinică mare în sindromul Waardenbur. În plus, Mckusick a asociat acest sindrom cu alte cursuri clinice similare, cum ar fi boala Hirschsprung..
În prezent, este considerată o patologie rară, care apare cu un grad variabil de insuficiență auditivă care poate provoca modificări semnificative ale învățării și dezvoltării ulterioare a persoanei afectate..
Prognosticul sindromului Waardenburg este favorabil, deși poate fi asociat cu morbiditate și mortalitate semnificative legate de complicații medicale, în special intestinale..
Sindromul Waardenburg este o tulburare genetică congenitală ale cărei semne și simptome tind să varieze mult între cei afectați..
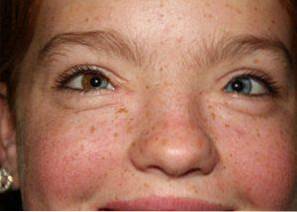
Cele mai frecvente trăsături includ anomalii faciale distinctive, pigmentare modificată a pielii, ochilor sau părului și surditate..
În literatura medicală, acest sindrom este adesea considerat un tip de genodermatoză sau neuropatie. Termenul de genodermatoză se referă la un set larg de boli caracterizate prin prezența unor anomalii ale pielii și modificări de origine genetică..
Pe de altă parte, termenul de neuropatie se referă la un grup de patologii derivate din dezvoltarea anomaliilor și a proceselor defecte în timpul migrației și diferențierii celulelor creastei neuronale în timpul gestației..
Cresta neuronală este o structură embrionară formată dintr-un set larg de celule nediferențiate a căror dezvoltare va duce la formarea structurii cranio-faciale și a celulelor neuronale și gliale care vor forma o mare parte a sistemului nervos..
Între a 8-a și a 10-a săptămână de gestație, începe de obicei procesul de migrare a celulelor care alcătuiesc creasta neuronală. Atunci când diferiți factori patologici sau anomalii genetice interferează în acest proces, pot apărea anomalii cognitive și / sau fizice semnificative, cum ar fi sindromul Waardenburg..
Prevalența sindromului Waardenbur este estimată la 1 caz la fiecare 40.000 de oameni din întreaga lume. De la descoperirea sa, aproximativ 1.400 de cazuri diferite au fost descrise în literatura medicală și experimentală..
Se pare că afectează în mod egal bărbații și femeile. Nu au fost identificate asociații cu regiuni geografice sau cu anumite grupuri etnice și rasiale.
Sindromul Waardenbug reprezintă 2-5% din toate cazurile diagnosticate de hipoacuzie congenitală.
Deși au fost identificate diferite cursuri clinice, tipurile I și II sunt cele mai frecvente. Tipul III și IV sunt rare.

Sindromul Waardenburg se caracterizează prin trei modificări fundamentale: alterări cranio-faciale, anomalii pigmentare și surditate:
O altă descoperire medicală centrală a sindromului Waardenburg este pierderea capacității auditive și a acuității. Cel mai frecvent este identificarea la cei afectați a unui grad variabil de surditate sau a pierderii auzului senzorial neural.
Termenul de hipoacuzie senzorială se referă la o pierdere a capacității auditive derivată din leziuni interne legate de terminațiile nervoase care conduc informații auditive de la urechea internă la centrele creierului..

Sindromul Waardenburg este clasificat în 4 tipuri de bază pe baza evoluției clinice și a simptomelor specifice care sunt prezente la persoanele afectate:
Sindromul Waardenbuug are o origine congenitală asociată cu diverse modificări genetice.
Analiza cazurilor a permis localizarea acestor anomalii în gene: EDN3, EDNRB, PAX3, SOX10, SNAI2 și MIT.
Acest set de gene pare a fi implicat în dezvoltarea și formarea diferitelor tipuri de celule, inclusiv a celor responsabili de producerea melanocitelor.
Melanocitele sunt responsabile pentru generarea melaninei, un pigment care contribuie la colorarea ochilor, a părului sau a pielii.
În funcție de diferitele cursuri clinice, putem identifica diferite modificări genetice:
După cum am subliniat în descrierea inițială, diagnosticul sindromului Waardenbug se face pe baza mai multor criterii majore și minore:
Pentru a stabili un diagnostic definitiv, este esențial să se identifice prezența a două criterii majore sau cel puțin unul major și două minore. În plus, este necesară utilizarea unor teste complementare: biopsie, audiometrie sau teste genetice.
Nu există nici un remediu pentru sindromul Waardenbug, deși pot fi utilizate abordări simptomatice..
Tratamentul celor mai frecvente semne și simptome necesită de obicei intervenția medicală din partea dermatologilor și oftalmologilor..
Pe de altă parte, în cazul tratamentului surdității senzoriale, un implant cohlear poate fi efectuat însoțit de o logopedie și o intervenție neuropsihologică..


Nimeni nu a comentat acest articol încă.