
Sindromul Williams este o tulburare de dezvoltare de origine genetică care este asociată cu un profil caracteristic al deficiențelor fizice și cognitive. Mai exact la nivel clinic, se caracterizează prin 4 puncte cardinale: 1) trăsături și caracteristici faciale atipice, 2) întârziere generalizată în dezvoltarea psihomotorie și profil neurocognitiv specific, 3) alterări cardiovasculare și t) posibilitatea dezvoltării hipercalcemiei infantile.
În ciuda faptului că sindromul Williams este considerat o boală rară, există mii de persoane afectate în întreaga lume. În ceea ce privește diagnosticul, examenul clinic oferă de obicei constatările necesare pentru stabilirea acestuia, cu toate acestea, pentru a exclude alte patologii și falsuri pozitive, se începe de obicei un studiu genetic prin diverse tehnici.
Pe de altă parte, nu există nici un remediu pentru sindromul Williams și nici un protocol de tratament standard, astfel încât majoritatea intervențiilor terapeutice vor încerca să regleze complicațiile medicale. În plus, va fi esențial să se includă în intervenții programe de îngrijire timpurie, educație specială individualizată și stimulare neuropsihologică..
Indice articol
Sindromul Williams este o tulburare de dezvoltare care poate afecta semnificativ diferite zone.
În general, această patologie se caracterizează prin prezența unor trăsături faciale atipice sau alterări cardiovasculare, dizabilitate intelectuală moderată, probleme de învățare și trăsături distinctive de personalitate..
Astfel, primul pacient cu sindrom Williams a fost descris de Dr. Guido Fanconi, într-un raport clinic din 1952. Cu toate acestea, cardiologul Joseph Williams a fost cel care în 1961 a identificat cu precizie această patologie, precum și a fost descris de germanul Beuren.
Datorită acestui fapt, sindromul Williams își primește numele de la ambii autori (sindromul Williams-Beuren), sau pur și simplu de la primul.
În ciuda faptului că, până acum câțiva ani, identificarea patologiei a fost efectuată pe baza caracteristicilor fenotipice, în 1993 Edward și colab. Au găsit o anomalie genetică în cromozomul 7q 11.23 ca fiind cauza etiologică..
Deși sindromul Williams este asociat cu o mare varietate de complicații medicale secundare, nu are o rată ridicată a mortalității. În multe cazuri, indivizii afectați sunt capabili să atingă un nivel funcțional independent.
Sindromul Williams este considerat o tulburare genetică rară sau rară.
Asociația Sindromului Williams, printre alte instituții, a estimat că sindromul Williams are o prevalență de aproximativ 1 caz la 10.000 de oameni din întreaga lume. Mai exact, s-a identificat că în Statele Unite pot exista aproximativ 20.000 sau 30.000 de afectați.
În ceea ce privește distribuția patologiei pe sexe, nu există date recente care să indice o prevalență mai mare la oricare dintre ele, în plus, nu au fost identificate diferențe între regiunile geografice sau grupurile etnice.
Pe de altă parte, știm, de asemenea, că sindromul Williams este o afecțiune sporadică, deși au fost descrise unele cazuri de transmitere a familiei.
Sindromul Williams, ca și alte patologii de origine genetică, are un curs clinic caracterizat prin implicarea multisistemică.
Mulți autori, precum González Fernández și Uyaguari Quezada, descriu spectrul clinic al sindromului Williams clasificat în mai multe domenii: caracteristici biomedicale, caracteristici psihomotorii și cognitive, caracteristici psihologice și comportamentale, printre altele..
Afectarea fizică prezentă în sindromul Wiliams este diversă, printre cele mai frecvente constatări clinice pe care le putem observa:
Dezvoltarea întârziată sau încetinită poate fi deja detectată în timpul sarcinii. Copiii afectați de sindromul Williams se nasc adesea cu greutate și înălțime reduse. În plus, odată ce stadiul adult este atins, înălțimea totală este de obicei mai mică decât cea a populației generale, aproximativ 10-15 cm.
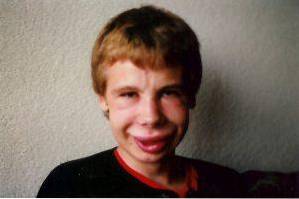
Modificările faciale sunt una dintre cele mai caracteristice descoperiri clinice în acest sindrom. La persoanele afectate putem observa o frunte semnificativ îngustă, pliuri cutanate marcate în fisura palpebrală, strabism, iris stelat, nas scurt și aplatizat, pomeți proeminenți și o bărbie mai mică decât de obicei..
În cazul alterărilor legate de dezvoltarea mușchilor și oaselor, este posibil să se observe prezența tonusului și forței musculare reduse, a laxității articulațiilor, a scoliozei, a contracturilor, printre altele. Vizual, se poate observa o postură caracterizată de umeri căzuți și extremități inferioare semi-flectate..
Deși de obicei nu există anomalii sau malformații semnificative în pinna auditivă, în toate cazurile se dezvoltă o creștere a sensibilității auditive. Persoanele afectate tind să perceapă sau să experimenteze anumite sunete ca fiind enervante sau dureroase.
Pielea are de obicei o elasticitate redusă, deci este posibil să se observe semne timpurii ale îmbătrânirii. În plus, herniile se pot dezvolta, în special în regiunea inghinală și ombilicală..
Diferitele anomalii ale inimii și ale vaselor de sânge constituie cea mai semnificativă complicație medicală, deoarece pot pune în pericol supraviețuirea persoanei afectate.
Dintre anomaliile cardiovasculare, unele dintre cele mai frecvente sunt stenoza aortică supravalvulară, stenoza ramurilor pulmonare, stenoza valvei aortice. Toate aceste modificări, la nivel clinic, pot afecta alte teritorii vasculare și chiar creierul, datorită dezvoltării hipertensiunii arteriale.
Anomaliile legate de funcția renală și vezica urinară sunt foarte frecvente. În plus, poate fi detectată și o acumulare de calciu (nefrocalcinoză), urgență urinară sau enurezis nocturnă.
La nivel cognitiv, cele mai semnificative caracteristici sunt constituite de o întârziere generalizată în dobândirea abilităților motorii, o întârziere intelectuală moderată și diverse modificări legate de percepția vizuală.
Sunt descrise diverse modificări legate de echilibrul și problemele de coordonare, care se datorează în principal prezenței anomaliilor musculo-scheletice și care vor provoca, printre altele, o întârziere în achiziționarea mersului, abilitățile motorii finale etc..
Este posibil să se găsească o întârziere mentală moderată, IQ-ul tipic al celor afectați oscilează de obicei între 60 și 70. În ceea ce privește zonele specifice care sunt afectate, există o asimetrie clară: pe lângă coordonarea psihomotorie, percepția și integrarea vizuală, de obicei să fie afectate în mod clar, în timp ce domenii precum limbajul sunt de obicei mai dezvoltate.
În etapele cele mai inițiale, există de obicei o întârziere în dobândirea abilităților lingvistice, cu toate acestea, de obicei, se recuperează în jur de 3-4 ani. Copiii cu sindrom Williams tind să aibă o comunicare expresivă bună, sunt capabili să utilizeze vocabular contextualizat, gramatică corectă, contact vizual, expresii faciale etc..
Una dintre cele mai semnificative descoperiri în sindromul Williams este comportamentul social excepțional al celor afectați. Deși în unele cazuri pot apărea crize de anxietate sau îngrijorări excesive, acestea sunt foarte empatice și sensibile.
Cele mai recente cercetări au indicat că cauza sindromului Williams se regăsește în diferite modificări genetice ale cromozomului 7. Cromozomii transportă informațiile genetice ale fiecărei persoane și se află în nucleul celulelor corpului..
La om, putem găsi 46 de cromozomi care sunt distribuiți în perechi. Acestea sunt numerotate de la 1 la 23, cu excepția ultimei perechi formate din cromozomii sexuali, numită XX în cazul femeilor, XY în cazul bărbaților. Astfel, în cadrul fiecărui cromozom poate exista o infinitate de gene.
Mai exact, procesul anormal identificat în sindromul Williams este o microcelecție sau descompunerea unei molecule de ADN care confirmă acest cromozom. În mod normal, acest tip de eroare are loc în faza de dezvoltare a gametilor masculi sau feminini..
Anomaliile genetice se găsesc în zona 7q11.23, în care au fost identificate peste 25 de gene diferite legate de tiparul clinic caracteristic al acestei patologii..
Unele gene, cum ar fi Clip2, ELN, GTF21, GTF2IRD1 sau LIMK1, sunt absente la cei afectați. Pierderea ELN este legată de țesutul conjunctiv, pielea și anomaliile cardiovasculare.
Pe de altă parte, unele cercetări indică faptul că pierderea genelor Clip2, GTF2I, GTF2IRD1 și LIMK1 poate explica modificări ale proceselor visuoperceptive, ale fenotipului comportamental sau ale deficitelor cognitive..
Mai mult, în mod specific, gena GTF2IRD1 pare să joace un rol proeminent în dezvoltarea trăsăturilor faciale atipice. La rândul său, gena NCF1 pare să fie legată de un risc ridicat de a dezvolta hipertensiune.
Până în ultimii ani, diagnosticul sindromului Williams se făcea exclusiv pe baza observării caracteristicilor fenotipice (alterări faciale, dizabilități intelectuale, deficite cognitive specifice, printre altele).
Cu toate acestea, în prezent, diagnosticul sindromului Williams se face de obicei în două etape: analiza constatărilor clinice și studii genetice de confirmare. Astfel, diagnosticul clinic include de obicei:
- Examinarea și evaluarea fizică și neurologică.
- Analiza parametrilor de creștere.
- Examinarea sistemului cardiorespirator.
- Examen nefrourologic.
- Analiza nivelurilor de calciu în urină și sânge.
- Analiza oftalmologică.
Pe de altă parte, analiza genetică este utilizată pentru a confirma prezența alterărilor genetice compatibile cu sindromul Williams, printre cele mai frecvente teste fiind tehnica de hibridizare in situ fluorescentă (FIHS)..
După extragerea unei probe de sânge, tehnica de hibridizare in situ se realizează prin marcarea sondelor ADN care sunt detectate sub o lumină fluorescentă..
Nu există un tratament specific pentru sindromul Williams, cu toate acestea, această patologie este asociată cu multiple complicații în diferite organe, astfel încât intervențiile medicale vor fi orientate spre tratamentul acestor.
Autorii González Fernández și Uyaguari Quezada subliniază că toate intervențiile trebuie să aibă un caracter multidisciplinar marcat, permițând tratamentul varietății simptomatice caracteristice acestui sindrom. În plus, acestea subliniază, de asemenea, diferite măsuri terapeutice în funcție de zona afectată:
În acest caz, complicațiile medicale, cum ar fi modificările cardiace sau malformațiile musculo-scheletice, necesită de obicei un tratament bazat în principal pe administrarea de medicamente și proceduri chirurgicale. Profesioniștii din diferite domenii (medici pediatri, cardiologi, oftalmologi etc.) participă de obicei la tratamentul simptomelor fizice..
Deficiențele cognitive, cum ar fi modificarea vizual-perceptivă sau întârzierea lingvistică, trebuie abordate încă din primele etape. Stimularea cognitivă și reabilitarea vor fi un factor determinant în realizarea unei vieți autonome în timpul maturității.
Deși cei afectați de sindromul Williams prezintă de obicei o bună funcționare socială, uneori au tendința de a prezenta comportamente excesiv de anxioase și de a dezvolta comportamente perseverente sau fobii.
Prin urmare, în aceste cazuri va fi esențial să se implementeze o abordare psihologică, prin diferite strategii care sunt eficiente pentru a minimiza aceste probleme sau dificultăți..



Nimeni nu a comentat acest articol încă.