Sindromul Smith-Lemli-Opitz este o tulburare metabolică care cuprinde mai multe simptome diferite, cum ar fi o creștere semnificativ lentă, trăsături faciale caracteristice, microcefalie, întârziere mentală ușoară sau moderată, dificultăți de învățare și probleme de comportament.
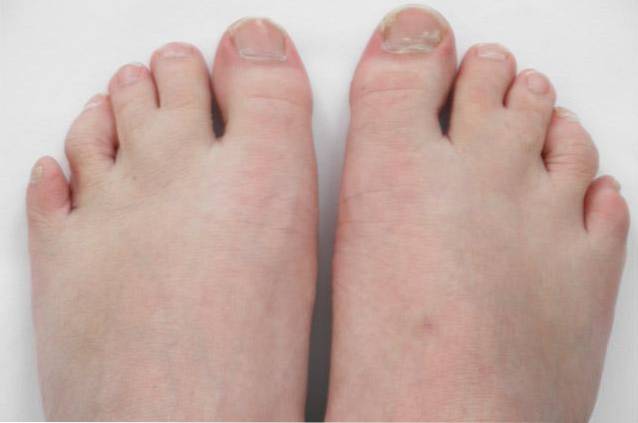
De asemenea, este însoțit de malformații la plămâni, inimă, rinichi, intestin și chiar la nivelul organelor genitale. În plus, pot prezenta sindactilie (fuziunea unora dintre degete) sau polidactilie (mai mult de 5 degete într-un picior sau o mână).
Se pare că cauza acestui sindrom este lipsa unei enzime care este importantă pentru metabolizarea colesterolului care este dobândit prin moștenirea genetică a unui model autosomal recesiv..
Cu toate acestea, aceste prezentări par să varieze enorm în funcție de gravitatea bolii chiar și în aceeași familie. Acest sindrom poate apărea în literatură cu nume precum deficiența de 7-dehidrocolesterol reductază, sindromul RSH sau sindromul SLO..
Indice articol
În 1964, medicii pediatri David Smith, Luc Lemli și Opitz John au descris 3 pacienți de sex masculin cu microcefalie și hipogenitalism și au definit această afecțiune ca RSH prin inițialele prenumelor originale ale acestor pacienți. Ulterior, numele sindromului a fost schimbat în numele de familie ale descoperitorilor.
Aproximativ 30 de ani mai târziu, Tint și colab. (1994) au constatat la 5 pacienți cu această afecțiune, concentrații semnificativ scăzute de colesterol în sânge, dar o creștere de peste 1000 de ori nivelurile de 7-dehidrocolesterol. Au văzut că această creștere se datorează lipsei unei enzime care ar trebui să transforme 7-dehidrocolesterolul în colesterol.
Ulterior, gena DHCR7 asociată cu această boală a fost identificată și clonată în 1998..
Sindromul Smith-Lemli-Opitz afectează aproximativ 1 din 20.000 până la 60.000 de nou-născuți vii din întreaga lume. De fapt, poate fi moștenit la 1 din 1590 până la 13.500 de indivizi, dar această cifră nu este utilizată, deoarece mulți fături cu această afecțiune mor înainte de naștere (Organizația Națională pentru Tulburări Rare, 2016).
În ceea ce privește sexul, acesta afectează în mod egal bărbații și femeile, deși este mai ușor diagnosticat la bărbați, deoarece malformațiile genitale sunt mai vizibile decât la femei.
Mai mult, pare a fi mai frecvent la persoanele de origine europeană; în special din țări aparținând Europei centrale precum Republica Cehă sau Slovacia. Cu toate acestea, este foarte rar în populația din Africa sau Asia..
Sindromul Smith-Lemli-Opitz apare din cauza mutațiilor genei DHCR7, prezentă pe cromozomul 11, care este responsabil cu trimiterea comenzilor de fabricare a enzimei 7-dehidrocolesterol reductază.
Aceasta este enzima care modulează producția de colesterol și ar fi absentă sau într-o măsură foarte mică în acest sindrom, ceea ce duce la o producție insuficientă de colesterol care ar împiedica creșterea normală..
Acest lucru are un impact mare, deoarece colesterolul este important în organism. Se compune dintr-o lipidă asemănătoare grăsimilor care se obține în principal din alimente de origine animală, cum ar fi gălbenușuri de ou, produse lactate, carne, carne de pasăre și pește..
Este esențial ca embrionul să se dezvolte fără dificultăți, având funcții importante, cum ar fi contribuirea la structura membranelor celulare și a mielinei (o substanță care acoperă celulele creierului). De asemenea, servește la producerea de hormoni și acizi digestivi.
Lipsa enzimei 7-dehidrocolesterol reductază determină acumularea în organism a unor componente potențial toxice ale colesterolului. Deci, avem, pe de o parte, niveluri scăzute de colesterol și, în același timp, acumularea de substanțe care pot fi toxice pentru organism; cauzând insuficiență de creștere, întârziere mintală, malformații fizice și probleme de organe interne.
Cu toate acestea, nu se știe cu siguranță completă cum aceste probleme asociate cu colesterolul dau naștere simptomelor sindromului Smith-Lemli-Opitz..
În prezent, peste 130 de mutații legate de sindrom au fost găsite în gena DHCR7, de fapt, există o bază de date care include toate cazurile descrise de sindrom Smith-Lemli-Opitz cu variantele lor, fenotipurile și genotipurile lor.
Deși există atât de multe mutații posibile, cele mai multe cazuri aparțin celor mai frecvente 5, iar restul sunt foarte rare.
Aceste mutații ale genei DHCR7 sunt moștenite cu un model autosomal recesiv, ceea ce înseamnă că o persoană care prezintă sindromul trebuie să fi moștenit gena mutantă de la ambii părinți. Dacă îl primiți de la un singur părinte, nu veți avea boala; dar ar putea fi un operator de transport și să-l transmită în viitor.
Există un risc de 25% ca ambii părinți purtători să aibă un copil afectat, în timp ce riscul ca copilul să fie purtător ar fi, de asemenea, de 50% în fiecare sarcină.
Pe de altă parte, în 25% din cazuri se poate naște fără aceste mutații genetice sau poate fi purtător; toate aceste date fiind independente de sexul bebelușului.
Trebuie luat în considerare faptul că există o probabilitate mai mare de a avea copii cu orice tulburare genetică recesivă dacă părinții care sunt rude apropiate (sau consanguine) decât părinții care nu au aceste legături.
Simptomele acestui sindrom variază în funcție de persoana afectată, în funcție de cantitatea de colesterol pe care o pot produce. Caracteristicile clinice acoperă mai multe aspecte și pot fi foarte diverse. Ele se găsesc în general pe față, membre și organe genitale; deși pot implica alte sisteme corporale.
Mulți dintre cei afectați au caracteristici tipice ale autismului, afectând interacțiunea socială. Dacă starea este ușoară, pot fi observate doar unele probleme de învățare și comportament; dar în cele mai grave cazuri, persoana poate avea o mare dizabilitate intelectuală și anomalii fizice care pot duce la moarte.
Există simptome care pot fi deja prezente de la nașterea individului, deși le vom include pe cele care apar în toate etapele vieții:
- Lipsa dezvoltării fizice observată după naștere.
- Întârziere mintală (100%).
- Microcefalie (90%).
- Sindactilie sau fuziune a 2 sau 3 degete (<95%).
- Ptoza pleoapelor, adică având una dintre pleoapele superioare căzute (70%).
- Meat urinar situat într-un loc diferit de cel normal la bărbați, cum ar fi în partea inferioară a glandului, trunchiului sau uniunii dintre scrot și penis. Este prezent în 70% din cazuri.
- Palatul despicat, care se manifestă ca un fel de gaură alungită în palat (50%).
- Foarte redusă maxilară sau micrognatie.
- Limba foarte mică (microglozie).
- Urechi scăzute.
- Nas mic.
- Coborâre incompletă a unuia sau a ambelor testicule.
- Hipotonie sau tonus muscular scăzut.
- Tulburari de alimentatie.
- Tulburări de comportament: comportamente antisociale, autodistructive și violente. Apar, de asemenea, comportamente de auto-stimulare tipice autismului, cum ar fi mișcările repetitive de legănare.
- Autism.
- Cataracta timpurie.
- Polidactilie sau încă un deget după degetul mic.
- Întârzierea creșterii în stadiul fetal.
- Organe genitale ambigue.
- Defecte cardiace.
- Rinichi multicistic.
- Absența unuia sau a ambilor rinichi la naștere.
- Boală de ficat.
- Hiperplazia suprarenală
- Anomalii pulmonare.
- Transpira excesiv.
- Anomalii cerebrale în structurile liniei medii, cum ar fi dezvoltarea incompletă a corpului calos, sept și vermis cerebelos.
- Acrocianoză: vasoconstricție cutanată care provoacă o culoare albăstruie în mâini și picioare.
- Picioare echinovare.
- Stenoza pilorică (15%)
- Boala Hirschprung, cauzând o motilitate intestinală slabă (15%)
- Fotosensibilitate.
- Înnorări sau comă.
- Acumularea de lichid în corpul fătului.
-Modificări în neurodezvoltare.
- Probleme neuropsihiatrice, care apar mai des atunci când ajung la maturitate.
- Respirație scurtă din cauza problemelor pulmonare.
- Pierderea auzului.
- Tulburări de vedere, care pot fi însoțite de strabism.
- Vărsături.
- Constipație.
- Convulsii.
Acest sindrom apare de la concepție, în ciuda faptului că, atunci când copilul se naște, simptomele nu sunt foarte clare și sunt mai subtile decât în copilăria târzie sau la maturitate; mai ales dacă sunt forme mai ușoare ale bolii. Din acest motiv, este detectat târziu în mai multe ocazii.
În orice caz, cel mai frecvent este că această afecțiune este deja suspectată la scurt timp după naștere din cauza malformațiilor pe care le prezintă de obicei..
Potrivit Organizației Naționale pentru Tulburări Rare, diagnosticul se bazează pe examene fizice și un test de sânge care detectează nivelul colesterolului. Este esențial ca copilul să fie evaluat pentru toate aspectele posibile asociate bolii, cum ar fi ochii, urechile, inima, mușchii scheletici, organele genitale și tulburările gastro-intestinale..
În ceea ce privește testele de sânge, un subiect cu sindrom Smith-Lemli-Opitz va avea o concentrație ridicată de 7-dehidrocolesterol (7-DHC) în sânge (un precursor care trebuie transformat de enzima 7-dehidrocolesterol reductază pentru a obține colesterolul) și niveluri foarte scăzute de colesterol.
Poate fi detectat și înainte de naștere printr-o tehnică cu ultrasunete sau ultrasunete, un dispozitiv care folosește unde sonore pentru a examina interiorul uterului femeii însărcinate. Cu această tehnică, pot fi observate deformările fizice tipice acestui sindrom..
Un alt test este amniocenteza, care constă în extragerea unei mici probe de lichid amniotic (unde se dezvoltă fătul) pentru a detecta defectele genetice. Aceleași informații pot fi obținute prin eșantionarea villusului corionic (CVS), extragând o probă de țesut din placentă..
Pe de altă parte, testele genetice moleculare pot fi utilizate pentru diagnostic prenatal, pentru a observa dacă există mutații în gena DHCR7 și dacă boala se va prezenta sau va fi doar un purtător.
Din păcate, majoritatea celor mai grave cazuri de sindrom Smith-Lemli-Opitz mor la scurt timp după naștere. Dacă există o dizabilitate intelectuală severă, este dificil pentru acești oameni să dezvolte o viață independentă.
Cu toate acestea, cu îngrijiri medicale adecvate și o dietă bună, acești pacienți pot duce o viață normală..
În prezent nu există un tratament specific pentru sindromul Smith-Lemli-Opitz. Acest lucru se datorează faptului că astăzi originea biochimică a bolii nu este cunoscută cu certitudine absolută, deoarece colesterolul are mai multe funcții complexe în metabolism.
Tratamentul medical pentru sindromul Smith-Lemli-Opitz se bazează pe problemele specifice găsite la copilul afectat și cel mai bine este să interveniți devreme.
Poate fi de mare ajutor pentru a primi suplimente de colesterol sau pentru a crește aportul acestuia prin dietă, pentru a îmbunătăți nivelul de dezvoltare și a reduce fotosensibilitatea. Uneori combinat cu acizi biliari.
Pentru intoleranță la soare, este recomandabil ca acești pacienți să folosească protecție solară, ochelari de soare și îmbrăcăminte adecvată atunci când ies afară..
S-a demonstrat că furnizarea de medicamente precum simvastatina poate reduce severitatea bolii. Deși, deoarece fenotipul clinic apare în timpul lipsei de colesterol în embriogeneză, acesta trebuie administrat în acel moment..
Pe de altă parte, un medicament antagonist al precursorului toxic al colesterolului care este în exces (7-dehidrocolesterol) poate fi, de asemenea, utilizat pentru a preveni creșterea acestuia. Suplimentele de vitamina E vă pot ajuta.
Alte tipuri de medicamente specifice pot fi utile pentru simptome precum vărsături, reflux gastroesofagian sau constipație..
Chirurgia sau aparatul dentar pot fi necesare dacă există deformări fizice sau probleme musculare legate de acest sindrom, cum ar fi fisura palatului, defecte cardiace, hipotonie musculară sau modificări genitale.
În concluzie, este necesar să se continue cercetările în acest sindrom, astfel încât să se dezvolte tratamente mai eficiente și specifice..


Nimeni nu a comentat acest articol încă.