facomatoză este un grup de tulburări neurocutane de origine genetică, rare la populația generală. La nivel clinic, acestea se caracterizează prin dezvoltarea unei implicări organice multisistemice cu leziuni cutanate sau tumorale, în diferite zone ale pielii, organelor sau sistemului nervos..
În plus, cursul său clinic nespecific îi face dificilă diagnosticul precoce, astfel încât consecințele sale medicale și psihologice deteriorează semnificativ calitatea vieții persoanei afectate și a rudelor acestora..
Deși există un număr mare de boli neurocutanate, cele mai frecvente includ fibromatoza de tip I și tip II, boala Bourneville, sindromul Sturge-Weber și boala Von Hippel-Lindau..
Pe de altă parte, în ciuda faptului că toate acestea sunt patologii congenitale, au fost concepute multiple abordări terapeutice de natură dermatologică care încearcă să îmbunătățească semnele și simptomele caracteristice acestor tulburări și, prin urmare, prognosticul medical al celor afectați..
Indice articol
Termenul de phakomatoză provine din expresia de origine greacă Phakos a cărui semnificație se referă la <
Patologiile neurocutanate se caracterizează fundamental prin existența unei asocieri semnificative între o afectare sau o tulburare neurologică și manifestările dermatologice.
Astfel, termenul de patologie neurocutanată este utilizat într-un mod generalizat pentru a cuprinde diferite boli care sunt prezente la persoana afectată congenital și care, în plus, pot fi prezente pe tot parcursul vieții odată cu dezvoltarea leziunilor cutanate și a tumorilor în diferite zone, sistemul nervos, sistemul cardiovascular, sistemul renal, sistemul cutanat, sistemul oftalmic etc..
În acest fel, termenul de phakomatoză a fost introdus în 1917 de Brouwer și mai târziu de van der Hoeve în 1923, cu toate acestea, descrierile inițiale făceau trimitere doar la unele patologii incluse în acest grup. În prezent, peste 40.
La nivel clinic, facomatoza este descrisă ca o boală care se prezintă cu alterări cutanate și malformații benigne / maligne în diferite sisteme: neurologice, oculare, cutanate și viscerale..
În ceea ce privește zonele afectate, diverși autori subliniază că cele de origine ectodermică sunt cele mai afectate, adică pielea și sistemul nervos, deși pot afecta și alte sisteme sau dispozitive, precum ochiul.
Sindroamele și patologiile de origine neurocutanată sunt boli rare în populația generală, deși nu există date specifice la nivel general despre toate acestea.
Astfel, epidemiologia acestor tulburări variază în funcție de tipul de boală, în special, neurofibromatoza este una dintre cele mai frecvente, cu o prevalență relativă de un caz la 300.000 de nașteri..
Bolile neurocutanate se caracterizează prin dezvoltarea leziunilor cutanate. Mai exact, facomatoza se distinge de multe altele prin prezența hamartomilor..
Hamartoamele sunt un tip de malformație benignă sau tumoră care poate crește în diferite organe, cum ar fi creierul, inima, ochii, pielea sau plămânii..
Cu toate acestea, facomatoza poate fi asociată cu un număr mare de afecțiuni medicale care vor varia, fundamental, în funcție de boala specifică sau de patologia suferită de persoana afectată..
În prezent, un număr mare de tulburări neurocutane au fost identificate la nivel clinic și genetic, cu toate acestea există unele cu o prevalență mai mare în populația generală: neurofibromatoza tip I și tip II, boala Bourneville, boala Von Hippel-Lindau și Sturge-Weber sindrom.
Există diferite forme clinice de neurofibromatoză. Cu toate acestea, în prezent cele mai frecvente sunt neurofibromatoza de tip I, numită și boala Von Reclinghausen și neurofibromatoza de tip II, urmată de shwannomatoza coloanei vertebrale.
La nivel etiologic, toate aceste manifestări medicale ale neurofibromatozei au o origine genetică și apar odată cu formarea tumorilor în zonele nervoase, în special în sistemul nervos central și periferic..
Formațiile tumorale, de obicei necanceroase sau benigne, tind să crească și să se dezvolte aproape oriunde în sistemul nervos, cum ar fi creierul, măduva spinării sau nervii periferici.
Astfel, algele de complicații medicale secundare la neurofibromatoză includ anomalii în creștere, dezvoltarea convulsiilor, apariția tumorilor cerebrale, patologii osoase, surditate și / sau orbire, sau dezvoltarea unor probleme semnificative de învățare, printre altele..
În plus, această patologie este prezentă din momentul nașterii. Cu toate acestea, manifestarea semnificativă a tabloului său clinic poate fi întârziată până la copilărie târzie, adolescență timpurie sau vârstă adultă..
Pe de altă parte, diagnosticul acestui tip de patologie include de obicei, pe lângă examenul fizic și neurologic, diferite teste de neuroimagistică și analize genetice..
În plus, în prezent nu există nici un remediu pentru neurofibromatoză, cu toate acestea, există abordări terapeutice specializate în controlul afectării dermatologice, acestea pot include atât tratamente farmacologice, cât și tratamente chirurgicale pentru oprirea sau eliminarea formațiunilor tumorale.
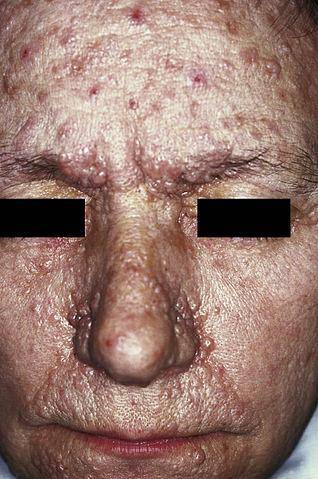
Neurofibromatoza de tip I (NF1), cunoscută și sub numele de boala von Recklinghausen, se manifestă în principal prin prezența unor pete maronii deschise, denumite în mod obișnuit „café au lait”, efelide (pistrui) și neurofibroame (leziuni ale nervilor în celulele Schwann și neurite).
Are o origine genetică autosomală dominantă, în special datorită unei mutații pe cromozomul 17, la locul 17q11.2. Astfel, gena implicată în
dezvoltarea neurofibromatozei de tip I, are un rol proeminent în modularea creșterii și diferențierii celulare și, în plus, poate funcționa ca un supresor tumoral.
În ceea ce privește epidemiologia acestei patologii, aceasta prezintă o prevalență aproximativă a unui caz pentru fiecare 2.500.3000 de nașteri.
Diagnosticul neurofibromatozei de tip I se face de obicei pe baza criteriilor clinice consensuale ale Institutului Național de Sănătate (1987), cu toate acestea, necesită o urmărire continuă pentru a evita complicațiile medicale secundare..
În mod normal, creșterile tumorale sunt tratate cu medicamente, pentru a preveni dezvoltarea exponențială a acestora sau prin îndepărtarea chirurgicală.
Neurofibromatoza de tip II (NF2), se manifestă în principal prin dezvoltarea schwanoamelor, adică formațiuni tumorale derivate din celulele Shcwaan care vor fi responsabile pentru acoperirea prelungirilor nervoase.
Schwannomele sau neuroamele afectează de obicei în special nervul auditiv și optic și într-o măsură mai mică zonele pielii.
Neurofibromatoza de tip II are o origine genetică autosomală dominantă, în special datorită prezenței unei mutații pe cromozomul 22, la locația 22q11.22.
Gena implicată în dezvoltarea acestei patologii este responsabilă pentru codificarea unei componente proteice cu rol proeminent în suprimarea tumorii, astfel încât activitatea sa deficitară produce o creștere anormală a proliferării celulare.
În ceea ce privește epidemiologia acestei patologii, este mai puțin frecventă decât tipul 1, prezentând o prevalență aproximativă a unui caz la 50.000 de nașteri.
Diagnosticul neurofibromatozei de tip II este similar cu tipul anterior și se face de obicei pe baza criteriilor clinice consensuale ale Institutului Național de Sănătate. Cu toate acestea, include de obicei teste de laborator complementare, cum ar fi neuroimagistica..
În mod normal, creșterile tumorale sunt tratate cu medicamente, cu toate acestea, în cazurile în care este posibil, se utilizează îndepărtarea chirurgicală.
Boala Bourneville este unul dintre termenii folosiți pentru a se referi la scleroza tuberoasă, o tulburare genetică caracterizată prin prezența hamartomilor.
La nivel clinic, poate da naștere unei afectări multisistemice caracterizate prin afectare cutanată (angioame faciale, fibroame unghiale, plăci fibroase, pete hipocromice etc.), afectare renală (angiomiolipome renale sau chisturi renale), afectare cardiacă (rabdomioame cardiace) , afectare neurologică (tuberculi corticali, noduli gliali subependimali, atrocitoame, convulsii, dizabilități intelectuale, anomalii comportamentale și motorii), printre altele.
La fel ca bolile descrise mai sus, originea sclerozei tuberoase este genetică. Mai exact, se datorează prezenței mutațiilor în genele TSC1 și TSC2..
Pe de altă parte, diagnosticul de scleroză tuberoasă se face pe baza criteriilor clinice propuse într-o conferință medicală din 1998. Cu toate acestea, studiul genetic este considerat relevant și pentru confirmarea sa.
În ceea ce privește tratamentul sclerozei tuberoase, deși nu există tratament, se utilizează de obicei diferite abordări farmacologice și chirurgicale, în principal pentru controlul creșterilor tumorale și a complicațiilor medicale secundare, cum ar fi manifestările neurologice.
Boala Von Hippel-Lindau, cunoscută și sub numele de angiomatoză retino-cerebeloasă, se manifestă în principal prin prezența și dezvoltarea malformațiilor vasculare, chisturi și / sau tumori, în general benigne..
Are o origine genetică autosomală dominantă, în special datorită unei mutații pe cromozomul 3, la locația 3p-25-26. În plus, prezintă o incidență estimată a unui caz la 40.000 de nașteri..
Mai exact, boala Von Hippel-Lindau afectează în principal sistemul nervos central (SNC) și retina, prin formarea hemangioamelor..
Hemangioamele sunt malformații vasculare caracterizate prin prezența grupurilor de capilare sanguine dilatate. Ele apar de obicei în zonele creierului și coloanei vertebrale, deși sunt frecvente și în retine sau pe piele.
Diagnosticul acestei patologii, pe lângă examenul fizic și neurologic, necesită un studiu oftalmologic detaliat, împreună cu analiza din diferite teste de neuroimagistică, pentru a confirma prezența leziunilor nervoase..
În ceea ce privește tratamentul bolii Von Hippel-Lindau, intervenția de bază este intervenția chirurgicală pentru eliminarea malformațiilor vasculare. Cu toate acestea, necesită monitorizare continuă pentru a evita complicațiile secundare..
În plus, are o speranță de viață redusă, în jur de 50 de ani, în principal datorită dezvoltării carcinoamelor cu celule renale (formațiuni neoplazice ale celulelor canceroase din tubii renali).
Sindromul Sturge-Weber, cunoscut și sub numele de angiomatoză encefal-trigeminală, se manifestă în principal prin prezența hemangioamelor.
Un hemangiom este un tip de neoplasm sau formare de tumori caracterizat prin prezența unui număr anormal de mare de vase de sânge în piele sau în alte organe interne..
Mai exact, la nivel clinic, sindromul Sturge-Weber se caracterizează prin dezvoltarea hemangioamelor faciale, hemangioamelor intracraniene și hemangioamelor coridice, conjunctivale, episcerale și glaucomului..
Are o origine genetică, în special datorită unei mutații pe cromozomul 9, la locația 9q21, în gena GNQ. Această componentă genetică are un rol proeminent în controlul factorilor de creștere, peptidelor vasoactive și neurotransmițătorilor (Orhphanet, 2014).
Diagnosticul sindromului Sturge-Weber se face pe baza suspiciunii clinice și a diferitelor teste de laborator, cum ar fi tomografia computerizată sau imagistica prin rezonanță magnetică..
Pe de altă parte, în ceea ce privește tratamentul, terapia cu laser este capabilă să reducă progresia acestei patologii și, în plus, în multe cazuri să elimine complet hemangioamele..



Nimeni nu a comentat acest articol încă.