
Sindromul DiGeorge este o patologie de origine genetică care se manifestă prin dezvoltarea malformațiilor legate de structura inimii, feței, timusului și glandelor paratiroide.
La nivel clinic, acestea vor produce o mare varietate de complicații medicale, printre care se numără deficiențele imune, hipocalcemia, patologiile cardiace și tulburările psihiatrice..

În ceea ce privește originea etiologică, aceasta este asociată cu o alterare genetică a cromozomului 22. Datorită acestui fapt, se mai numește sindrom de deleție 22q11.2..
Diagnosticul se bazează pe identificarea semnelor clinice cardinale prin examinarea fizică și diferite teste de laborator: examen analitic și imunologic, ecografie abdominală, ecocardiograme și studiu genetic, bazat fundamental pe hibridizare fluorescentă in situ (FISH)..
În cele din urmă, tratamentul acestei patologii se concentrează pe corectarea malformațiilor organice și controlul complicațiilor medicale. Astfel, se folosesc de obicei terapia cu limfocite T, suplimentele de calciu, chirurgia corectivă etc..
Indice articol
Această patologie a fost descrisă inițial de specialistul pediatric american Angelo M. DiGeorge în 1965. În raportul său clinic, DiGeroge a descris o patologie congenitală definită de dezvoltarea deficientă sau absența glandei paratiroide și a timusului.
Ulterior, Chapelle din 1918 a descris în mod specific defectele congenitale derivate din această patologie. Astfel, sindromul DiGeorge a fost denumit a doua cauză a defectelor cardiace congenitale după sindromul Down.
În cele din urmă, această patologie a fost caracterizată clinic prin triada clasică de imunodeficiență, endocrinopatie cu hipocalcemie și boli de inimă..
Mai mult, în multe cazuri, heterogenitatea simptomatică largă a delețiilor localizate pe cromozomul 22 implică diferențierea a trei tipuri diferite de patologii la nivel clinic:
- Sindromul DiGeorge
- Sindromul velocardiofacial
- Sindromul cardiofacial
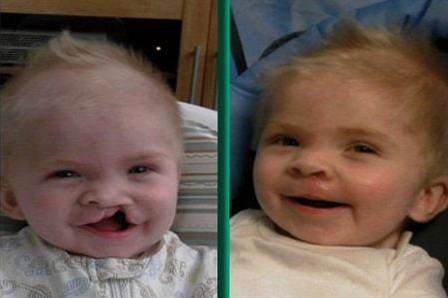
Sindromul DiGeorge, cunoscut și sub numele de sindromul de deleție 22q11.2, este o boală cauzată de un defect genetic care are ca rezultat dezvoltarea diferitelor malformații organice și organice.
În acest sens, acest sindrom derivă fundamental din procesele de dezvoltare defectuoase din faza prenatală sau de gestație, localizate în principal în a treia și a 8-a săptămână de gestație..
Mai exact, în jurul celei de-a cincea săptămâni de gestație, structurile embrionare încep un proces de formare și dezvoltare a diferitelor structuri și organe (Vera de Pedro și colab., 2007).
Astfel, un grup de anumite celule va da naștere dezvoltării feței, a diferitelor părți ale creierului, timusului, inimii, aortei și glandelor paratiroide..
Acest „câmp de celule” este de obicei localizat în jurul zonei sau zonei din spatele gâtului embrionului în gestație. În acest fel, pentru ca restul structurilor să înceapă să se formeze și să se diferențieze, este esențial ca aceste celule să se deplaseze către diferitele zone specifice pentru fiecare structură..
În această fază de dezvoltare, se formează bursa faringiană, arcurile și fisurile, timusul și glandele paratiroide și mai târziu, o parte a structurilor craniene și faciale sau diferite porțiuni ale țesutului conjunctiv..
În acest fel, anomaliile genetice tipice sindromului DiGeroge dau naștere unei modificări sistematice a acestui proces de formare prenatală, provocând eșecuri grave ale dezvoltării.
În consecință, zonele cele mai afectate sunt de obicei:
- Inima: această structură constituie unul dintre organele vitale pentru supraviețuirea noastră. Face parte din sistemul circulator și funcția sa esențială este de a pompa sânge către restul corpului.
- Configurare facială: formarea structurii faciale depinde de formarea corectă a craniului, globilor oculari, sistemului bucal, urechilor etc..
- Înșelătorie: această structură joacă un rol fundamental în cadrul sistemului imunitar, deoarece este responsabilă pentru maturizarea limfocitelor sau a celulelor T.
- Glande paratiroide: sunt constituite de un set de glande endocrine care au un rol mizat în reglarea calciului, printre alți factori.
Astfel, zonele cele mai afectate în sindromul DiGeorge sunt legate de defectul formării embrionare în zonele asociate cu gâtul și regiunile adiacente..
Sindromul DiGeroge are o prevalență estimată de 1 caz la 4.000 de persoane din populația generală.
Cu toate acestea, numeroase studii epidemiologice indică o prevalență mai mare, în principal datorită eterogenității cursului său clinic și dificultății stabilirii unui diagnostic precoce..
În plus, atât în Statele Unite, cât și pe plan internațional, sindromul DiGeorge este considerat una dintre cele mai frecvente cauze de defecte cardiace congenitale și malformații faciale..
Pe de altă parte, în ceea ce privește caracteristicile epidemiologice de natură sociodemografică, a fost identificată o prevalență de 1 caz la 6.000 de persoane de origine caucaziană, asiatică și afro-descendentă, în timp ce în cazul hispanilor, prevalența se ridică la un caz pentru la fiecare 3.800 de indivizi.
În cazul celor mai frecvente semne și simptome în sindromul DiGeorge, trebuie să subliniem că acesta prezintă un curs clinic cu expresivitate variabilă.
În acest caz, la unii pacienți complicațiile medicale prezintă o stare severă, care poate duce la moarte timpurie. În alte cazuri, caracteristicile prezintă de obicei un compromis minim pentru supraviețuirea și funcționalitatea persoanei afectate..
Prin urmare, nu toți cei afectați de sindromul Di George vor prezenta aceeași afecțiune, cu toate acestea, de obicei includ una sau mai multe modificări asociate.
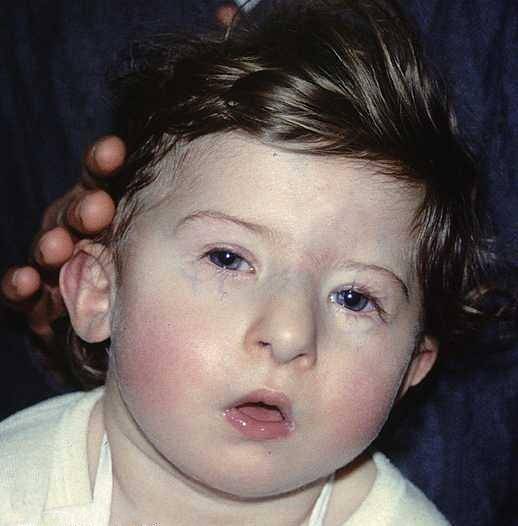
Modificările legate de configurația feței constituie una dintre cele mai izbitoare trăsături vizuale ale sindromului DiGeorge, în general acestea sunt definite de:
- Microcefalie: capul se dezvoltă cu o dimensiune mai mică sau mai mică decât se aștepta pentru nivelul de dezvoltare și vârsta cronologică a persoanei afectate. În plus, o structură nazală tubulară se dezvoltă de obicei însoțită de obraji plati sau slab accentuați.
- Hploplazie și retrognatie mandibulare: structura maxilarului nu este complet dezvoltată. Astfel, în multe cazuri are o dimensiune redusă sau o poziție modificată, situată mai în spate decât de obicei..
- Alterarea oculară: în general, ochii tind să fie incluși spre planul inferior, în plus, în jurul ochilor pot apărea microftalmie (subdezvoltarea unuia dintre globii oculari), cataractă (opacitatea cristalinului ocular) sau cianoză (colorare albastră).
- Modificarea pinnei: este posibil să se identifice o asimetrie în configurația urechilor. De obicei au o implantare scăzută cu prezența malformațiilor în lobi și în alte zone exterioare ale pinnei.
- Malformații orale: configurația gurii prezintă de obicei un aspect arcuit spre planul superior, caracterizat prin prezența unui sul nazolabial lung și accentuat și a palatului despicat.
Anomaliile cardiace includ adesea o mare varietate de defecte. Cu toate acestea, cele mai afectate zone sunt legate de aorta și structurile cardiace asociate:
- Defecte septale: peretele sau structura care separă camerele cardiace responsabile de pomparea sângelui, pot fi formate incomplet sau defectuos.
- Malformația arcului aortic: Diverse anomalii pot fi descrise și în segmentul aortic situat între căile ascendente și descendente.
- Tetralogia lui Fallot: această patologie se referă la prezența alterărilor defectului septal ventricular, îngustarea semnificativă a arterei pulmonare, poziția anormală a aortei și îngroșarea zonei ventriculare drepte.
Persoanele afectate de sindromul DiGeorge au de obicei o susceptibilitate semnificativă la contractarea diferitelor tipuri de patologii, în principal infecțioase (viruși, ciuperci, bacterii etc.).
Acest fapt se datorează prezenței unei disfuncții a sistemului imunitar, datorită unei dezvoltări deficitare a tipului și a producției de limfocite și celule T.
Sistemul imunitar este alcătuit dintr-o mare varietate de organe, structuri, țesuturi și celule care împreună ne protejează de agenții patologici interni și de mediu..
În acest sens, sindromul DiGeorge produce o formare deficitară sau incompletă a timusului, ducând la modificări ale funcționalității și localizării sale finale..
În general, cea mai proeminentă anomalie este hipofuncționalitatea limfocitelor T, esențială în producerea de imunoglobuline și anticorpi..
În acest caz, persoanele afectate de sindromul Digeorge au de obicei niveluri anormal de scăzute de concentrație de calciu în organism și în sânge..
Această afecțiune derivă fundamental din prezența anomaliilor în glandele paratiroide, din cauza unei subdezvoltări a componentelor sale (PrimaryInmune, 2011).
Aceste glande sunt situate în gât și se află într-o poziție apropiată de tiroidă. Cu toate acestea, în acest caz au un volum redus, deci va avea un impact semnificativ asupra controlului metabolismului și echilibrului calciului din organism..
Astfel, în acest caz, nivelul de calciu din sânge este de obicei sub 2,1-8,5 mm / dl, provocând diferite complicații medicale precum crampe, iritabilitate musculară, amorțeală, schimbări de dispoziție, deficite cognitive etc..
Pe lângă semnele și simptomele descrise mai sus, este posibilă identificarea altora legate de sfera cognitivă și intelectuală a celor afectați..
În special în cazurile diagnosticate, au fost descrise dificultăți de învățare, deficit intelectual moderat, deficit de atenție, modificări ale dispoziției, tulburări de anxietate, printre altele..
Originea genetică a sindromului DiGeorge este asociată cu prezența alterărilor în cromozomul 22, în mod specific în locația 22q11.2. Mai exact, se datorează absenței unei secvențe de ADN, alcătuită dintr-un număr de 30 până la 40 de gene diferite..
Deși o bună parte a genelor implicate nu au fost încă identificate în detaliu, absența acestui grup mare apare în mai mult de 90% din cazuri ca mutație de novo, în timp ce aproximativ 7% se datorează factorilor ereditari.
Pentru stabilirea diagnosticului sindromului DiGeorge, este esențial să se identifice semnele clinice cardinale ale acestei patologii:
- Defecte faciale.
- Defecte cardiace.
- Imunodeficiență.
- Hipocalcemie.
În acest sens, împreună cu analiza istoricului medical și a examenului fizic, este esențial să se efectueze diferite teste de laborator precum ecocardiografie, ultrasunete, examen imunologic și studii analitice serice..
În plus, un aspect important este examenul genetic, acesta fiind realizat în principal prin hibridizare fluorescentă in situ (FISH).
După cum am subliniat în descrierea inițială, tratamentul este destinat în principal controlului și corectării semnelor și simptomelor cauzate de acest tip de boală..
În cazul hipocalcemiei, se tratează de obicei prin administrarea de suplimente de calciu și / sau vitamina D..
Pe de altă parte, în cazul deficienței imune, deși tind să se îmbunătățească odată cu înaintarea în vârstă, pot fi utilizate diferite abordări, cum ar fi transplantul unei părți a țesutului timusului, terapia cu limfocite T sau transplantul de măduvă osoasă..
În ceea ce privește malformațiile faciale și orale, se folosesc de obicei reparații chirurgicale, care îmbunătățesc aspectul fizic și funcționalitatea acestor os.
În cele din urmă, în cazul modificărilor cardiace, ambele medicamente pot fi administrate pentru tratamentul și corectarea lor prin intervenție chirurgicală..
În majoritatea cazurilor, persoanele afectate ajung de obicei la maturitate, cu toate acestea, un procent semnificativ dintre acestea începe să dezvolte anomalii imunologice și / sau cardiace importante care cauzează moartea prematură, în special în primul an de viață..



Nimeni nu a comentat acest articol încă.