
Sindromul Joubert este o tulburare de origine genetică care se caracterizează printr-o scădere a tonusului muscular, probleme de coordonare, mișcări anormale ale ochilor, modificări ale modului de respirație și dizabilitate intelectuală (Fundația Sindrom Joubert, 2016).
Toate aceste modificări se datorează unei transmisii genetice autozomale care va provoca anomalii cerebrale importante, reducerea vermisului cerebelos, precum și anomalii în structura trunchiului cerebral (Institutul Național pentru Tulburări Neurologice și Accident vascular cerebral, 2016).
În plus, sindromul Joubert face parte dintr-un grup de tulburări numite ciliopatii care implică o disfuncție a unei părți a celulelor numite cili. Fundația Sindrom Joubert, 2016).
Descrierea inițială a acestei patologii a fost făcută de Marie Joubert și colab. În 1968, în care au fost descrise patru cazuri. La pacienți a existat absența parțială sau totală a vermisului cerebelos, sindromul neonatal de ampnee-hipernee episodică, mișcări anormale ale ochilor, ataxie și retard mental (Angemi și Zucotti, 2012).
În plus, acest sindrom a fost, de asemenea, asociat cu diferite modificări multiorganice, cum ar fi fibroza hepatică, polidactilia, nefronoptizia sau distrofia retinei (Angemi și Zucotti, 2012).
În ceea ce privește tratamentul, în prezent nu există un remediu pentru sindromul Joubert. Intervențiile terapeutice vizează controlul și sprijinul simptomatic, stimularea fizică și intelectuală a copiilor și terapia ocupațională (Institutul Național pentru Tulburări Neurologice și Accident vascular cerebral, 2016).
Indice articol
Sindromul Joubert (JS) este un tip de patologie de origine genetică care se caracterizează printr-o malformație congenitală în zonele trunchiului cerebral și agenezie (absență parțială sau completă) sau hipoplazie (dezvoltare incompletă) a vermisului cerebelos, care poate provoca (Ophatnet , 2009).
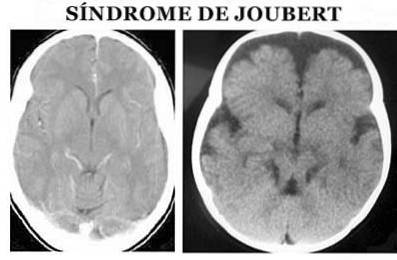
Mai precis, la nivel anatomic se caracterizează prin așa-numitul semn molar al creierului mediu: agenezie sau hipoplazie a vermisului cerebelos, îngustarea pedunculilor cerebeloși superiori cu îngroșare, alungire și lipsă de decutație și fosă interpedunculară profundă (Angemi și Zuccoti, 2012).
Este o tulburare care poate afecta multe zone și organe ale corpului, astfel încât semnele și simptomele variază considerabil în rândul persoanelor afectate (Biblioteca Națională de Medicină din SUA, 2011).
Majoritatea celor afectați suferă de tonus muscular slăbit (hipotonie) și dificultăți de coordonare motorie (Ataxia). Alte trăsături caracteristice sunt: episoade de respirație modificată, nistagmus (mișcare involuntară și aritmică a ochilor), dezvoltare motorie întârziată și dificultăți intelectuale variabile (Biblioteca Națională de Medicină din SUA, 2011).
Prevalența sindromului Joubert a fost estimată la aproximativ 1 / 80.000 până la 1 / 100.000.000 de nașteri vii. La nivel mondial, au fost înregistrate peste 200 de cazuri clinice (Angemi și Zuccoti, 2012).
Mulți specialiști consideră că aceste cifre sunt subestimate, deoarece sindromul Joubert are o gamă largă de afecțiuni și este subdiagnosticat (Biblioteca Națională de Medicină din SUA, 2011).
Multe dintre simptomele clinice ale sindromului Joubert sunt mai mult decât evidente în copilărie, mulți copii afectați au întârzieri motorii semnificative (Organizația Națională pentru Boli Rare, 2011).
Cele mai frecvente caracteristici ale evoluției clinice sunt: lipsa controlului muscular (ataxie), modificarea modelelor de respirație (hipercapnie), apnee în somn, mișcări anormale ale ochilor (nistagmus) și tonus muscular scăzut (Organizația Națională pentru Boli Rare, 2011).
Pe de altă parte, unele dintre modificările care pot fi asociate cu sindromul Joubert includ: dezvoltarea alterată a retinei, anomalii ale irisului, strabism, rinichi și / sau alterări hepatice, proeminența membranelor care acoperă creierul, printre altele ( Organizația Națională pentru Boli Rare, 2011).
Toate modificările derivate din acest sindrom sunt cuprinse în mai multe domenii: alterări neurologice, oculare, renale și musculo-scheletice (Bracanti și colab., 2010).
Cele mai caracteristice alterări neurologice ale sindromului Joubert sunt Bracanti și colab., 2010): hipotonie, ataxie, întârziere generalizată în dezvoltare, alterări intelectuale, alterarea modelelor respiratorii și mișcări anormale ale ochilor.
La nivel fizic, retina este unul dintre organele afectate de sindromul Joubert. Modificările din acest organ apar sub formă de distrofie retiniană, datorită unei degenerări progresive a celulelor responsabile de recepția foto..
Din punct de vedere clinic, modificările oculare pot varia de la orbirea congenitală a retinei până la degenerescența progresivă a retinei.
Pe de altă parte, este posibilă și observarea prezenței colobomului. Această alterare oculară este un defect congenital care afectează irisul ocular și apare ca o gaură sau o fantă.
Patologiile legate de funcția renală afectează mai mult de 25% dintre cei afectați de sindromul Joubert.
În multe cazuri, anomaliile renale pot rămâne asimptomatice timp de câțiva ani sau pot începe să se manifeste cu semne nespecifice, până când se prezintă ca insuficiență renală acută sau cronică..
Din primele descrieri ale acestei patologii, o constatare clinică frecventă este polidactialia (o tulburare genetică care crește numărul degetelor sau de la picioare).
În plus, este, de asemenea, obișnuit să observăm anomalii orofaciale sau structurale la nivelul coloanei vertebrale.
Studiile experimentale au clasificat sindromul Joubert drept o tulburare autosomală recesivă (Organizația Națională pentru Boli Rare, 2011).
O tulburare genetică autozomală recesivă înseamnă că trebuie să existe două copii ale unei gene anormale pentru ca trăsătura sau boala să fie prezentă (National Institutes of Health, 2014).
Prin urmare, o modificare genetică recesivă apare atunci când o persoană moștenește aceeași genă anormală pentru aceeași trăsătură de la fiecare părinte. Dacă o persoană primește doar o copie a genei legate de boală, va fi purtător, dar nu va prezenta simptome (Organizația Națională pentru Boli Rare, 2011).
În plus, cel puțin zece gene au fost identificate ca fiind una dintre cauzele posibile ale sindromului Joubert (Organizația Națională pentru Boli Rare, 2011).
O mutație a genei AHI1 este responsabilă de această afecțiune patologică la aproximativ 11% din familiile afectate. La persoanele cu această alterare genetică, modificările vederii sunt frecvente datorită dezvoltării distrofiei retinei (Organizația Națională pentru Boli Rare, 2011).
Mutația genei nphp1 este cauza a aproximativ 1-2% din cazurile de sindrom Joubert. La persoanele cu această modificare genetică, modificările renale sunt frecvente (Organizația Națională pentru Boli Rare, 2011).
Pe de altă parte, o mutație a genei CEP290 este cauza a 4-10% din cazurile de sindrom Joubert (Organizația Națională pentru Boli Rare, 2011).
În plus, mutațiile genelor TME67, JBTS1, JBTS2, JBTS7, JBTS8 și JBTS9 sunt, de asemenea, legate de dezvoltarea sindromului Joubert (Organizația Națională pentru Boli Rare, 2011).
Diagnosticul sindromului Joubert se face pe baza simptomelor fizice. Este necesar să se efectueze atât o examinare fizică detaliată, cât și utilizarea diferitelor teste de diagnostic, în special a imaginilor cu rezonanță magnetică (Ophatnet, 2009).
În plus, testele genetice moleculare sunt adesea utilizate pentru a identifica modificările genetice care au fost demonstrate în 40% din cazurile de sindrom Joubert (Organizația Națională pentru Boli Rare, 2011).
Pe de altă parte, este de asemenea posibil să se facă un diagnostic prenatal al acestei patologii prin ultrasunete fetale și analize moleculare, în special în familiile cu istoric genetic al sindromului Joubert (Ophatnet, 2009).
Când cele mai caracteristice trăsături ale sindromului Joubert apar în combinație cu una sau mai multe patologii fizice suplimentare, se poate face un diagnostic al sindromului Joubert și al tulburărilor conexe (JSRD) (Biblioteca Națională de Medicină din SUA, 2011).
Prin urmare, în funcție de tipul de patologie asociată cu prezența sindromului Joubert, putem găsi subtipuri ale acestuia. Cu toate acestea, sistemul de clasificare a sindromului Joubert este încă într-o fază de evoluție datorită descoperirii contribuțiilor genetice și cunoașterii mai mari a corelațiilor fenotipice..
Prin urmare, putem găsi (Bracanti și colab., 2010):
Tratamentul utilizat în sindromul Joubert este simptomatic și susține patologiile de bază. Pe lângă intervențiile farmacologice, este obișnuit să se utilizeze stimularea fizică și cognitivă timpurie (Institutul Național pentru Tulburări Neurologice și Stoke, 2016).
Atunci când modificările respiratorii sunt semnificative, în special în fazele inițiale ale vieții, este necesară monitorizarea funcției respiratorii (Institutul Național al Tulburărilor Neurologice și Stoke, 2016).
Pe de altă parte, identificarea și controlul degenerescenței oculare, complicațiilor renale și restul complicațiilor legate de sindromul Joubert ar trebui efectuate cât mai curând posibil pentru a ajusta măsurile terapeutice (Institutul Național al Tulburărilor Neurologice și Stoke, 2016 ).

Nimeni nu a comentat acest articol încă.