
sindromul Potter este o tulburare moștenită autozomală recesivă rară și gravă care afectează nou-născuții și se caracterizează prin oligohidramnios marcat (lipsa lichidului amniotic), rinichi polichistic, agenezie renală și uropatie obstructivă.
Această boală a fost descrisă pentru prima dată de patologul Edith Potter în 1946, care a subliniat trăsăturile faciale similare ale unei serii de copii care aveau agenezie renală bilaterală. De acolo, a dezvăluit treptat simptomele tipice ale bolii..
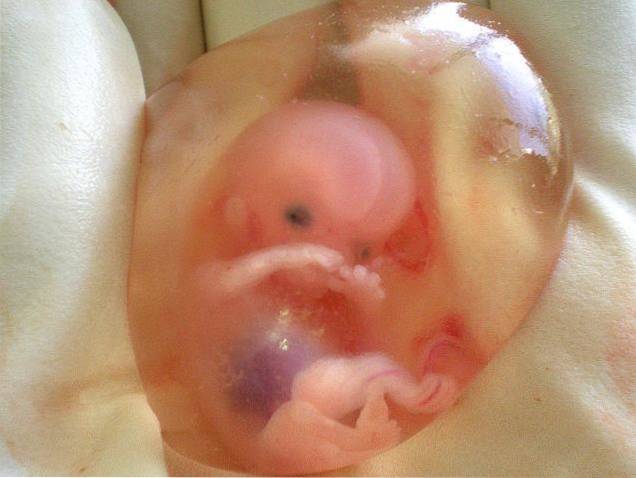
De asemenea, a fost numită secvența Potter sau secvența oligohidramnios. Conceptul sindromului Potter se referea la început numai la cazurile cauzate de agenezie renală bilaterală, deși în prezent mulți cercetători îl folosesc pentru orice caz care apare asociat cu lipsa lichidului amniotic..
Indice articol
Sindromul Potter apare la aproximativ 1 din 4000 de născuți și făt, cu toate acestea, există date recente care estimează că frecvența poate fi mult mai mare..
Bărbații au mai multe șanse decât femeile să dezvolte acest sindrom. Acest lucru se poate datora ratei mai mari la bărbații cu abdomen prune (sau boala Eagle-Barrett) și uropatiei obstructive (boli care sunt asociate cu acest sindrom). Deși s-a suspectat că cromozomul Y are ceva de-a face cu el. Oricum, acest lucru nu se știe cu siguranță..
Bebelușii născuți cu acest sindrom mor de obicei foarte devreme sau sunt născuți mort. Acest lucru se datorează de obicei insuficienței pulmonare și ageneziei renale bilaterale..
33% dintre copii mor în uter, în timp ce o rată de supraviețuire de 70% a fost documentată la 23 de sugari cu sindrom Potter și hipoplazie pulmonară.
Nou-născuții cu cea mai ușoară formă de sindrom Potter pot avea complicații din insuficiența respiratorie tipică, pneumotorax și insuficiență renală acută. Cei care ajung la prima copilărie pot avea boli pulmonare cronice și insuficiență renală.
Producția de urină la făt este principalul mecanism pentru a produce un volum adecvat de lichid amniotic, care începe în jurul celei de-a patra luni de sarcină. Fătul înghite continuu lichid amniotic, acesta este re-absorbit în intestin și apoi este expulzat din nou prin rinichi (în urină) în cavitatea amniotică.
În această boală, cantitatea de lichid amniotic este insuficientă, în principal deoarece rinichii bebelușului nu funcționează bine. În mod normal, ceea ce se întâmplă este că, în perioada de gestație, rinichii nu se formează corespunzător, lipsind unul sau ambii (ageneză renală).
Deși poate apărea și obstrucție a tractului urinar sau, uneori, ruptură a membranei care cuprinde lichidul amniotic. Această lipsă de lichid amniotic este principala cauză a simptomelor sindromului Potter..
Boala Potter poate apărea din două boli genetice, care sunt ambele boli renale polichistice autosomale dominante și autosomale recesive. În acest fel, un istoric familial de boli de rinichi poate crește riscul de a dezvolta acest sindrom la făt..
Astfel, în cazurile de familii cu antecedente de agenezie renală unilaterală sau bilaterală, aceasta poate fi o trăsătură autosomală dominantă..
Deși anumite mutații genetice au fost asociate cu afecțiuni frecvent observate în sindromul Potter, cum ar fi bolile renale polichistice autosomale recesive sau dominante și displazia renală multicistică, nu se găsește nimic definitiv în agenezia renală bilaterală..
Pe scurt, trăsăturile genetice specifice nu sunt cunoscute cu certitudine astăzi și este ceva care continuă să fie investigat.
Ceea ce se știe este că nu pare să existe o asociere directă a abuzului de substanțe sau a factorilor de mediu periculoși în timpul sarcinii cu apariția ageneziei renale bilaterale sau a sindromului Potter..
Principalele simptome ale acestui sindrom sunt:
- Principalul defect în secvența Potter este insuficiența renală.
- Lipsa lichidului amniotic: care poate provoca multe probleme deoarece lichidul ajută la lubrifierea părților corpului fătului, îl protejează și contribuie la dezvoltarea plămânilor săi. Când acest lichid lipsește, cavitatea amniotică este mai mică decât în mod normal și ajunge să lase puțin spațiu pentru făt, ceea ce împiedică creșterea sa normală.
- Naștere prematură
- Malformații: în special la extremitățile inferioare, cum ar fi picioarele și arcuirea picioarelor. De asemenea, poate apărea sirenomelia sau sindromul sirenei, care constă în fuziunea picioarelor.
- Aspect facial atipic, cum ar fi o punte nazală largă sau un nas de „papagal-cioc”, ochi mari și urechi așezate mai jos decât în mod normal.
- Excesul de piele, cu o pliere a pielii în zona obrazului fiind frecventă la cei afectați.
- Glandele suprarenale cu aspect de mici discuri ovale care apasă pe abdomenul posterior asociate cu disfuncționalitate renală.
- Vezica mai mică decât în mod normal și nu foarte dilatabilă, stocând foarte puțin lichid.
- La bărbați pot lipsi vasele deferente și veziculele seminale.
- La femei este posibil ca uterul și partea superioară a vaginului să nu se dezvolte.
- Atrezia anală: apare atunci când rectul nu este conectat corect la anus. La fel se poate întâmpla și în esofag, duoden sau artera ombilicală.
- Uneori poate apărea o hernie diafragmatică congenitală care împiedică dezvoltarea corectă a diafragmei.
- Plămânii imaturi sau hipoplazia pulmonară (anomalie congenitală caracterizată printr-o întrerupere a dezvoltării pulmonare conform Tortajada și colab., 2007). Acest mecanism nu este complet clar, deși pare să influențeze mișcarea corectă a lichidului amniotic prin plămâni în timpul etapei fetale. Evident, dacă nu există suficient lichid amniotic, plămânii nu se vor dezvolta corespunzător.
- În consecință, la cele de mai sus, există probleme respiratorii grave care sunt de obicei cauza mortalității timpurii la cei afectați.
În plus față de cele menționate deja, sindromul Potter a fost legat de alte probleme, cum ar fi sindromul Down, sindromul Kallmann și sindromul branchial-oto-renal (BOR), printre altele..
În timpul sarcinii se poate observa prin ultrasunete dacă există mai puțin lichid amniotic decât este necesar sau dacă fătul prezintă anomalii ale rinichilor sau absența acestora.
Pentru a detecta posibile probleme la nou-născut, poate fi necesară o radiografie a plămânilor și a abdomenului..
Pe de altă parte, puteți merge la un consilier genetic care va lua o probă de sânge de la făt pentru a efectua o amniocenteză. Acesta este folosit pentru a vedea dacă numărul de cromozomi este corect sau dacă există modificări în unele dintre părțile sale sau translocații.
Acest lucru poate fi util în excluderea altor boli asociate, cum ar fi sindromul Down. Pentru a detecta posibile mutații moștenite, explorarea genomului tatălui, mamei, bebelușului afectat și fraților este esențială..
Nu există tratamente pentru această boală și prognosticul acesteia este foarte negativ, de obicei mor înainte de naștere sau la scurt timp. Dacă supraviețuiește la naștere, poate fi necesară resuscitarea. Unele metode pot fi, de asemenea, utilizate pentru ameliorarea simptomelor și îmbunătățirea vieții cât mai mult posibil, cum ar fi transplantul de organe sau intervenția pentru uropatia obstructivă.
Cu toate acestea, există un caz al unui copil cu sindrom Potter născut în iulie 2013, expus de Jaime Herrera Beutler care trăiește astăzi. Acest lucru se datorează faptului că cu câteva săptămâni înainte de naștere, o soluție salină a fost injectată în uterul mamei cu scopul de a ajuta dezvoltarea pulmonară fetală..
Când s-a născut bebelușul, s-a constatat că intervenția fusese un succes și că poate respira singură. Ultimele știri pe care le avem despre ea sunt publicate pe 15 aprilie 2016 și supraviețuiește după ce a suferit un transplant de rinichi.



Nimeni nu a comentat acest articol încă.